Software Support
The Samuel J. Wood Library provides a wide array of licensed scientific software for use at Weill Cornell Medical College.
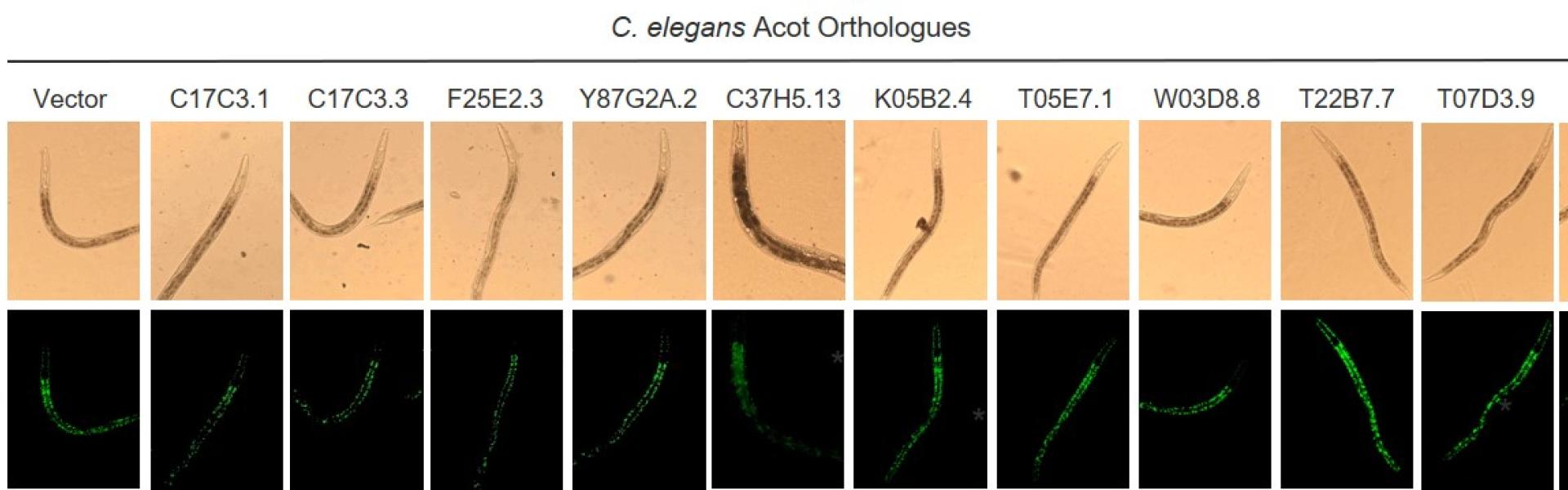

Obesity results in metabolic and cardiovascular diseases (CVD) due to inefficient utilization of excess lipids and sugars in energy expenditure, which become channeled toward cytotoxic pathways instead. The utilization of all lipids and sugars in metabolism requires their breakdown into acetyl-CoA within the mitochondria. However, obesity-induced maladaptive mechanisms that suppress channeling of excess acetyl-CoA toward energy expenditure remain unexplored. In Ersoy Lab, we aim to identify new mechanisms that channels acetyl-CoA away from energy expenditure and promote obesity and metabolic disorders.
In a recent publication in Hepatology, we demonstrated that obesity initiates a maladaptive hepatic response whereby excess lipids are deactivated by the acyl-CoA thioesterase (Acot) family members. Using RNAi screen in C. elegans, we identified Acot9 as the strongest regulator of lipid accumulation among the 15 known Acot family members. We went on to outline the mechanisms by which the expression of Acot9 within the liver promotes hepatic steatosis and glucose production by trafficking acetyl-CoA toward the anabolic pathways of the citric acid (TCA) cycle. This article was covered by a complimentary editorial and highlighted on the cover of the journal. Our ongoing studies are geared toward identifying the mechanisms by which Acot9 expression in extrahepatic tissues reduces the bioavailability of acetyl-CoA for energy expenditure. Our newly acquired preliminary data indicates that Acot9 expression within the brown and beige adipose tissue (BAT) limits thermogenesis, which blocks the consumption of excess energy as heat, and by doing so, promotes weight gain and hyperlipidemia
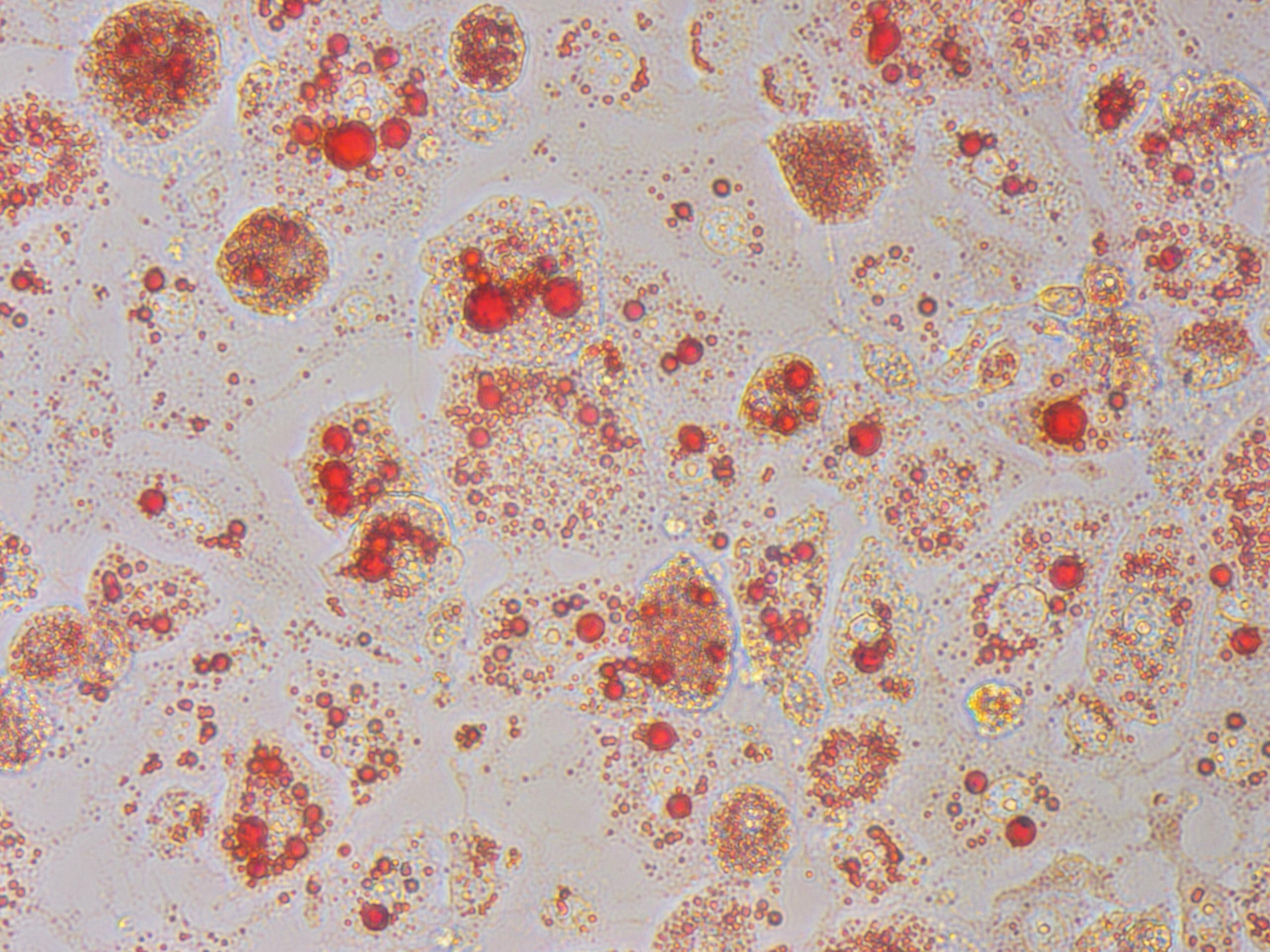
The pathogenesis of type 2 diabetes mellitus and nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitis (NASH) is linked to obesity-induced hepatocellular stress responses such as endoplasmic reticulum and oxidative stress. Because current treatment options are limited, the identification of novel mechanisms that regulate hepatic stress is critical to address this unmet demand.
We work on identifying maladaptive stress responses to excessive feeding, which contribute to the pathogenesis of metabolic disorders. We built a target discovery platform by monitoring the obesity-induced changes in the nuclear localization pattern of proteins in response to nutritional state using tandem-mass-tag proteomics. By mining this data set, Ersoy lab unveiled new paradigms as well as several new candidate proteins that are either exacerbated or impaired in the setting of metabolic disease. In the lab, we use CRISPR-Cas9 technology to knockdown candidate nuclear factors to test their potential as drug target for the treatment of metabolic disorders. In our early studies, Y box-binding protein 1 (Ybx1) exhibited the strongest nuclear enrichment following diet-induced obesity. Increased Ybx1 activity is associated with tumor metastasis and drug-resistant cancers; however, a biological role in the pathogenesis of metabolic disorders remains unexplored. We initially used Crispr-Cas9 systems, which was followed by transgenic mouse models, to demonstrate that hepatic Ybx1 activity contributes to diet-induced increases in lipids and glucose in the liver as well as the circulation. The feasibility of Ybx1 as a drug target for the treatment of diabetes, NAFLD and CVD is currently underway.

Steensels S, Qiao J, Zhang Y, Maner-Smith KM, Kika N, Holman CD, Corey KE, Bracken WC, Ortlund EA, Ersoy BA. Acot9 traffics mitochondrial short-chain fatty acids towards de novo lipogenesis and glucose production in the liver. Hepatology. 2020;72(3):857-872
“Copyright © 2000-2021 by John Wiley & Sons, Inc. or related companies. All rights reserved.”
The Samuel J. Wood Library provides a wide array of licensed scientific software for use at Weill Cornell Medical College.

We utilize complementary in vivo and in vitro models as well as human tissue in our research.